The case of Adrenochrome in autism spectrum disorders
Several small-scale studies conducted in the 1950s and 1960s reported that oxidized adrenaline metabolites, e.g. adrenochrome and adrenolutin, triggered psychotic reactions such as thought disorder and derealization when administered to healthy volunteers. We present adrenochrome and adrenolutin’s biochemical properties, and their possible role in higher cortical dysfunction and hypothesize them to be key triggers for the core symptoms observed in patients affected with autism spectrum disorder (ASD). An integrative model is presented.
A shift from enzymatic inactivation of adrenaline towards autoxidation into adrenochrome suggests that excess free radicals, xenobiotics, and genetic single-nucleotide-polymorphisms are contributing to the expression of symptoms observed in ASD. Several chemicals, e.g. silver oxide and aluminum salts catalyze adrenochrome’s oxidation to the psychoactive metabolite adrenolutin. Adrenochrome’s receptor affinity involves the central α2c adrenoceptor system, proposed to be dysregulated in several psychiatric diseases, e.g. schizophrenia. α2c receptor signaling reduces the hyperpolarization activated cation current, leading to an increased firing in dopaminergic cell groups. Emerging studies suggest not only clinical, but also biological links between ASD and schizophrenia. We propose that adrenochrome and adrenolutin are the culmination of oxidative stress and excess adrenaline. A brief review of key mechanisms is presented. Further studies quantifying these aminochromes in patients affected with ASD and neurotypical controls are warranted.
BACKGROUND
We begin our discussion with the integration of ASD with an open disclaimer. To focus attention on the implications of adrenochrome and adrenolutin in the pathophysiology of ASD, a neglected area in ASD research, the discussion presented here focuses on a specific hypothesis. Thus, this perspective is not aimed to be a comprehensive review of the entire literature of autism research, but rather is focusing on the plausible role of adrenochrome (and adrenolutin) in the etiology of this increasingly prevalent condition. Adrenochrome is formed by the autoxidation of adrenalin. Various reasons for elevations in adrenaline, e.g. genetic polymorphisms, excess dietary sugars and chemicals combined with oxidative stress will favor adrenaline’s autoxidation into adrenochrome and adrenolutin. These aminochromes have been demonstrated to have psychomimetic properties. We propose they induce altered perceptions in children affected with ASD, due to excess dopamine release in the prefrontal cortex and striatum by activating the alpha-2c-adrenoceptor system, leading to various deficits and clinical disease.
Adrenaline
At the turn of the 19th century a powerful physiologically active molecule was isolated from the human adrenal gland [132]. The structure of this physiologically active substance was described as beta-hydroxy-beta-(3,4-dihydroxyphenyl)-N-methylethylamine and was given the name epinephrine (Greek: epi-nephros = onto the kidney) (Fig 1). It was liberated from the benzoate by saponification with dilute sulfuric acid. Around the same time, other researchers isolated this active molecule as well and named it adrenaline (Latin, ad-renes = towards the kidneys) [1]. As a frequency analysis of the use of the two names in the titles and abstracts of bioscientific publications and national pharmacopoeias show, the name adrenaline is preferred to epinephrine in most parts of the world. Hence, throughout this review we will use the term adrenaline [126].
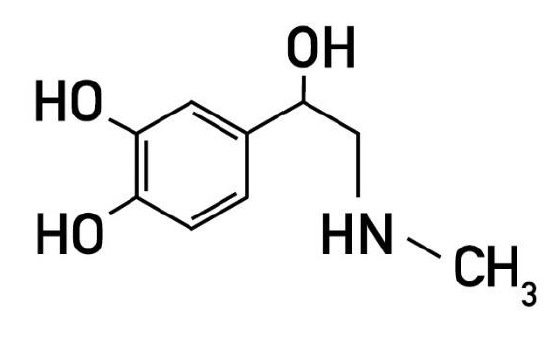
Figure 1 – Adrenaline [i.e., beta-hydroxy-beta-(3,4-dihydroxyphenyl)-N-methylethylamine
Central and Peripheral Catecholamine Metabolism
Adrenaline’s rapid inactivation explains the transience of its effects. Roughly 70% of peripheral and central adrenaline is deactivated by catechol-O-methyltransferase (COMT) to metanephrine. Besides COMT, any remaining adrenaline is metabolized via deamination by monoamide oxidase (MAO) as well as sulfate esterase and adrenaline oxidase. A small amount of adrenaline, however, is inactivated by quinoid autoxidation to the pink and fluorescent green colored aminochromes, e.g. adrenochrome and adrenolutin, respectively [Fig.2]. Both prompt a broad spectrum of activity [8].
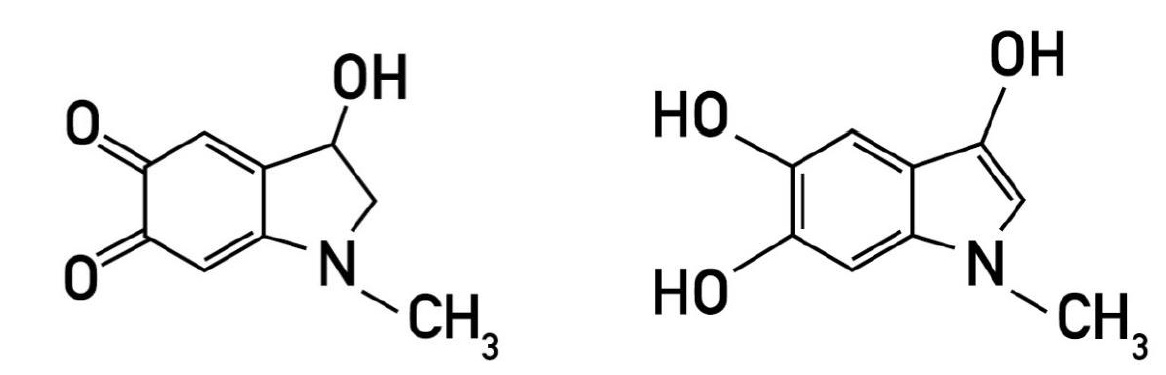
Figure 2 – I: Adrenochrome [i.e., 2, 3-dihydro-3-hydroxy-N-methyIindole-5,6-quinone], II: Adrenolutin [i.e., 3,5,6-trihydroxy-N-methylindo 3,5,6-trihydroxy-N-methylindole].
Suboptimal Catecholamine Metabolism
In the central nervous system (CNS) COMT is the principal enzyme responsible for the degradation of catecholamines, e.g. dopamine, as well as the monoamine neurotransmitters, e.g. epinephrine, norepinephrine, serotonin, and tyramine. Hence, polymorphic variations in COMT would suggest a shift from adrenaline’s enzymatic deactivation to increased oxidation and formation of adrenochrome and adrenolutin.
The gene coding for COMT is located on chromosome 22q11 and is primarily expressed in the prefrontal cortex (PFC) and hippocampus [133]. The COMT Val158Met (rs4680) polymorphism is a substitution of amino acid valine to methionine. This change affects dopamine regulation in the PFC. It has been reported that COMT polymorphism is involved in schizophrenia, autism, bipolar disorder, obsessive-compulsive disorder, hyperactivity, diseases such as migraine and in the pathogenesis of aggressive and antisocial behavior [128-129,134-137]. A meta-analysis of 12 studies demonstrated that there was a significant relationship between the COMT gene Val158Met genotype and executive function in healthy individuals [127].
There is growing evidence that the gene causing the symptoms of 22qDS, e.g. DiGeorge syndrome, may be that encoding COMT. The activity of dopamine in the brain is normally terminated by reuptake by a transporter protein. This protein is relatively inactive in the prefrontal cerebral cortex, suggesting that it is here that catechol-O-methyltransferase has the primary responsibility for the inactivation of dopamine [138].
Mice deprived of the ability to produce COMT tend to confirm this because they develop elevated dopamine levels but only in the prefrontal cortex [139].
Besides COMT polymorphisms, lack of signaling molecules further attenuate the genetic tone in regards to enzymatic catecholamine metabolism. S-adenosyl-L-methionine (SAMe) is a methyl donor for COMT, e.g. facilitating its expression and thereby promoting enzymatic adrenaline metabolism. SAMe synthesis involves methylenetetrahydrofolate reductase (MTHFR) activity. A meta-analysis studying the association of MTHFR 677C>T and 1298A>C polymorphisms demonstrated an increased risk of autism overall [130]. This suggests that reduced synthesis of SAMe would result in hypomethylation and would attenuate overall enzymatic catecholamine metabolism driven by COMT further. These complex relationships make it overall difficult to quantify the clinical relevance of single polymorphic relationships, as other genes, e.g. MTRR, MTR, MAT1A, MTHFD1, FOLR2, DHFR, DF and their respective (e.g. lack of) signaling molecules could further contribute to the blunted enzymatic metabolism in this patient population.
Finally, oxidation of adrenaline can be stimulated by both by chemicals. In fact, alkalis, metallic cations, various oxidizing agents and reactive oxygen species such as superoxide anion, hydrogen peroxide, hydroxyl radical [2-4] and nitric oxide together with its derivatives [5-7] have been demonstrated to stimulate the oxidation of catecholamines.
Adrenochrome
Green and Richter enzymatically oxidized adrenaline with tyrosinase and discovered an unstable red-crystalline aminochrome formed in the early stages of the reaction, e.g. 2,3-dihydro-3-hydroxy-l-methylin-dole-5,6-quinone, and named it adrenochrome [14]. The oxidation is rapid and involves electron removal, largely in reactive oxygen species (ROS) generating systems (e.g. superoxide, O2•-). It was demonstrated that superoxide dismutase inhibited the autoxidation of adrenaline when measured at pH 10.2 [15]. Superoxide catalyzes the conversion of adrenaline to adrenochrome by acting as propagating anion in a cascade reaction during the univalent oxidation of this catecholamine. Adrenochrome formed within cells can undergo conjugation with cellular glutathione (GSH), leading to its depletion, or polymerize into several other compounds, e.g. adrenolutin [16].
In 1956, it was first reported that a patient affected with asthma used an expired adrenaline spray and subsequently suffered from changes in perception, mood and thought [9]. It was subsequently confirmed that oxidized adrenaline, e.g. adrenochrome, created behavioral changes in various animals and was psychotomimetic when administered intravenously to healthy volunteers [9-10]. The effects of adrenochrome and adrenolutin in humans have been confirmed in subsequent publications and involved lack of insight, body image disturbances, paranoia, anxiety, thought disorder, as well as bizarre and inappropriate behaviors [11-12]. Since the pioneering studies of Green and Richter [14], the chemistry of adrenochrome has received attention in relation to the postulated role of this substance in psychiatric disorders, e.g. schizophrenia. A spectrophotometric method analyzing the in vitro oxidation of adrenaline in schizophrenic serum in fact demonstrated that it oxidized faster [13].
Adrenolutin
The formation of highly colored oxidation products from adrenochrome was reported in the early investigations into the chemistry of this substance [8]. The attempted purification of crude adrenaline oxidation mixtures was reported to give rise to the formation of the strong yellow-green fluorescence substance 3,5,6-trihydroxy-N-methylindo-3,5,6-trihydroxy-N-methylindole, e.g. adrenolutin, in several independent studies [17-19].
Adrenolutin administered to healthy volunteers often created prolonged psychiatric symptoms. Some of these symptoms have been described in case reports, with symptoms reportedly sustained for up to seven days [10].
These observations rapidly led to the development of empirical assay methods for these physiologically active compounds. Present-day chemical estimations of catecholamines and the correlating aminochromes are based on high-performance liquid chromatography (HPLC) coupled to triple quadrupole mass spectrometer systems.
Although neither adrenochrome nor adrenolutin have been measured and compared between schizophrenic, non-schizophrenic and normal control groups of the population, it is a reasonable hypothesis among other competing hypotheses of the schizophrenia syndrome.
Interestingly, aluminum salts, as well as other inorganic salts were shown to catalyze the oxidation from adrenochrome to adrenolutin [20-22]. It is known that aluminum salts are used as vaccine adjuvants in the United States. The quantities of aluminum present in vaccines are regulated by the Center for Biologics Evaluation and Research (CBER). The FDA recommends a preterm infant should not exceed 5 micrograms per kilogram of the baby’s weight per day, as greater exposures are associated with central nervous system and bone toxicity [140]. According to the 2019 CDC vaccination schedule, by 18 months a child receives a total of 4925 micrograms of injected aluminum [141]. In a recent small study involving autism brain autopsies, very high levels of aluminum were detected in every case [142]. If these levels of aluminum could contribute to higher formations of adrenolutin in patients affected with ASD remains to be proven.
Adrenochrome’s proposed influence on the Adrenoceptor System in Neuropsychiatric Disease
Adrenochrome and adrenolutin unexpectedly demonstrated selective receptor affinity for the central alpha-2c-adrenoceptor system (AR) [23].
α2a- and α2c-adrenoceptors (ARs) are the primary α2-AR subtypes involved in central nervous system (CNS) function. These receptors are implicated in the pathophysiology of psychiatric illness, particularly those associated with affective, psychotic, and cognitive symptoms [24].
Although 90% of α2-ARs in the CNS are contributed by the α2a-AR, the expression of the α2c-AR is more restricted, constituting approximately 10% of the total [25]. This suggests the possibility of significant differences in how these two receptor subtypes modulate regional neurotransmission.
All α2-AR subtype mRNAs were found to be expressed in rat brain but showed marked regional heterogeneity, e.g. the receptor specific density of distribution. α2c-ARs densely populate the ventral and dorsal striatum, hippocampus and the ventral tegmental area (VTA) in humans and monkeys [26-29,122]. Interestingly, the highest densities of α2c-adrenoceptors in rodents are found in the striatum and the olfactory tubercles [30].
The α2- (and α1) ARs specifically appear to play a prominent role in modulating prefrontal cortex (PFC) tone and as such mediate the effect of normal, aroused, and stressed neurotransmitter levels on memory and other cognitive processes [31]. The α2c-AR seems to play a key role in the dysregulation observed in neuropsychiatric disease [24]. Dopaminergic (DA) neurons have been shown to express α2c receptors [32]. Just recently, it was discovered that the α2c receptors reduce the hyperpolarization activated cation current, leading to an increased firing in DA cells [33]. The DA hypothesis implicating noradrenergic dysfunction has significant support in the literature [34]. The prefrontal cortex (PFC), striatum, and hippocampus are implicated in schizophrenia and ASD, where noradrenergic and dopaminergic terminals expressing a2c-receptors are represented.
Information derived from the cortex is transformed through the VTA and conducted to the striatum, where representations of motor and cognitive action sequences can be integrated and developed into more complex units for implementing efficient behavioral responses [38]. This provides strong evidence that, in addition to modulating motor control and motor learning, the striatum plays an important role in the formation of complex behaviors, including decision making and executive function [39].
While α2a-AR has been shown to be the predominant subtype in the brain and the major regulator of presynaptic inhibition of catecholamine release [40-41], and the expression level of α2c-ARs is very low in the medial prefrontal cortex, it has been suggested that α2c-AR is the main adrenergic modulator of the firing of dopamine cells in the ventral tegmental area (VTA) [42], where the prefrontal dopaminergic nerves originate. The VTA sends its 2 primary efferent fibers to the prefrontal cortex as well as the nucleus accumbens (NAc), an integral part of the striatum [123]. Interestingly, just in the last 2 years it was demonstrated elegantly that children affected with ASD have reduced structural connectivity in the mesolimbic pathways linking the NAc and VTA, two brain areas critical for processing social reward [131].
Adrenochrome provoked dopamine release in the prefrontal cortex may suggest it to be a pulse-like phenomenon fueled by peripheral adrenaline surges and redox imbalances leading to adrenochrome driven VTA hyperactivation.
Various single photon emission computed tomogram (SPECT) techniques were utilized to assess brain perfusion in patients affected with ASD and demonstrated prefrontal cortex hypoperfusion in every subject affected [120-122]. Increased plasma C-reactive protein (CRP) levels were associated with decreased connectivity between the ventral striatum and ventromedial prefrontal cortex, which in turn correlated with increased anhedonia [124]. Interestingly, elevated CRP levels during pregnancy increased the risk of ASD by 43% [125].
Experiments with selective α2c-AR antagonists, e.g. JP-1302, ORM-10921, and ORM-12741 consistently show improved pre-pulse inhibition (PPI) in Sprague Dawley and Wistar rats in NMDA-antagonist-induced models of schizophrenia [35-37].
Adrenochrome Hypothesis in Autism Spectrum Disorder (ASD)
Historically, autism spectrum disorders (ASD) and schizophrenia were intimately related [43-46]. In the early 20th century, Swiss psychiatrist Paul Bleuer first used the term autism [43]. He described it as a symptom of schizophrenia. Austrian-American psychiatrist Leo Kanner, however, independently characterized autism and schizophrenia, mainly due to the divergent onset of the conditions. Kanner’s original reports described autistic children behaving as if they were hypnotized. It wasn’t until 1980, however, until the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) separated schizophrenia and autism. The strong phenotypic relationship, however, suggests overlapping disease mechanisms involved in ASD and at least a subset of schizophrenia patients [47]. Behavioral phenotypes shared between autism spectrum disorders and schizophrenia prepare the ground for biological pathway analyses [47], as they share multiple phenotypic similarities and risk factors and have been reported to co-occur at elevated rates [56]. Interestingly, elevations in noradrenaline and adrenaline are reported in patients with ASD [48-52] as well as schizophrenia [53-55]. Cognitive symptoms of schizophrenia include deficits in explicit memory, as well as deficits in attention, working memory, and executive function [58-59], congruent with symptoms observed in ASD [60-63].
In the early stages of human life, central nervous system homeostasis is critical. Interference with neural circuitry, cell migration and exuberant synaptogenesis changes the trajectory of normal cognitive development. In ASD, the observed symptoms define the condition, as patients often have difficulties with proper communication. In contrast, characteristic negative symptoms of schizophrenia, e.g. delusions and hallucinations, are described by the actual patient, under clinical guidance. The differentiation of these conditions was hence warranted, as the approach to diagnose these two patient groups required discrete strategies. The last four decades, however, have provided an abundance of emerging evidence with much needed insight into the deeper biological relationship of ASD and schizophrenia, contributing to the blurring of phenotypic and genetic boundaries between these two disorders.
Autism Spectrum Disorder: An Integrative Model
ASD is a behaviorally defined neurodevelopmental disorder and generally recognized to have a complex etiology involving both genetic and environmental factors [116-117]. While the precise mechanisms underpinning its etiology are yet to be determined, research suggests genetic and environmental factors to be involved. The findings to date implicate a pattern of increased arousal to acute stress rather than persistent hyperarousal in many children with ASD [64-65]. These findings underscore the importance of perceived threat when we analyze the concept of acute stress. We suggest that individuals affected with ASD inherited this altered perception due to adrenochrome’s aberrant signaling of the α2c-AR system.
If adrenochrome and adrenolutin are in fact key molecules involved in core characteristic phenotypic expressions observed in autism spectrum disorder, then understanding and preventing its synthesis and enhancing its metabolism seems imperative. Epinephrine surges are suggested to be rapidly autoxidized by superoxide free radicals and hydrogen peroxide into the quinone adrenochrome and adrenolutin [15-16 and 66-67]. We propose an attenuated genetic tone in response to various epigenetic challenges responsible for the negative symptoms in ASD. Core mechanisms involved in an integrative model of autism are described suggesting ASD to be a chronic condition fueled by adrenochrome, where a group of elements is required to be present to favor its formation, e.g. excess free radicals, adrenaline, xenobiotics, and glucose.
Oxidative Stress in ASD
Many food-borne protein exorphins and toxins that can interfere with immune, oxidant, and neurological systems in humans were found in the fluids of children with ASD [69-77]. ASD and other neurodevelopmental disorders are related to increased endotoxins in plasma [78]. Xenobiotics may increase gut permeability, damage intestinal functions, and increase macromolecule adsorption, eventually overwhelming the system’s antioxidant capacity, generating redox imbalances and ultimately oxidative stress.
Oxidative stress in response to environmental exposure plays a role in many human diseases and is also presumed to be involved in the etiology of ASD [79-92]. To date, more than 115 publications reported oxidative stress to be associated with ASD [93]. Glutathione acting in cooperation with its dependent enzymes is known as the glutathione system and is responsible for the detoxification of reactive species and electrophiles produced by xenobiotics [94]. Glutathione occurs in two forms ‒ the oxidized form (glutathione disulfide or GSSG) and the reduced form (GSH) [95]. Glutathione is the most abundant endogenous antioxidant in the brain [96]. Its antioxidant function is mediated by reacting with reactive oxygen species (ROS), reactive nitrogen species (RNS), hydroxyl radicals (OH•),hypochlorous acid (HOCl), and other reactive species, or by acting as an indispensable cofactor for numerous enzymes especially glutathione peroxidases (GSH-Pxs) and glutathione S-transferases (GSTs) [97].
Patients affected with ASD are thought to be more vulnerable to oxidative stress because of their imbalance in intracellular and extracellular glutathione (GSH) levels and decreased glutathione reserve capacity. Several studies have suggested that the redox imbalance and oxidative stress are integral parts of ASD pathophysiology. The glutathione to oxidized glutathione ratio (GSH:GSSG) was found to be lower in children with ASD [98]. Oxidative stress decreases the GSH:GSSG ratio [99-100] and increases free radical generation in ASD [100]. The significant increase in plasma GSSG levels indicates that these children are under oxidative stress.
Superoxide dismutase (SOD) is an enzyme that catalyzes the dismutation (or partitioning) of superoxide (O2−) radical into ordinary molecular oxygen (O2) and hydrogen peroxide (H2O2). SOD levels were found to be lower in patients affected with ASD [101].
One possible source of excess free radicals is suggested to be the gastrointestinal system. The prevalence of GI symptoms in children with ASD varies from 9-91% [102]. The most comprehensive meta-analysis to date revealed that children with ASD were more than four-fold more likely to develop GI problems than those without ASD and, further, that constipation, diarrhea, and abdominal pain are reported most [103].
Diet and Sugar
A recent meta-analysis reported ASD patients exhibit food aversions, habitual eating behaviors as well as food selectivity (45% vs 17% in all children). These dietary behaviors can contribute to the development of nutrient deficiencies (such as deficiencies of vitamins, minerals, and fatty acids) and further fuel core autistic symptoms [104-106].
An interesting observation is the rise in plasma insulin, which tended to be greater in subjects receiving incremental doses of adrenaline [107]. In a large meta-analysis of maternal factors linked to autism, gestational diabetes was associated with the greatest increase (twofold) in the incidence of ASD. Although insulin does not cross the placenta, the elevation of fetal blood glucose levels due to maternal diabetes is predicted to increase fetal insulin secretion [108].
Present findings show that the consumption of a high-energy cafeteria diet (equivalent to the ‘western diet’) potentiated the ability of adrenaline to increase circulating glucose levels. Adrenaline injection attenuated insulin-stimulated removal of glucose from the bloodstream, thereby extending the duration in which circulating glucose concentrations remain elevated while on a western diet. The insulin-resistance was reversed in 4 weeks by returning to the baseline diet [109]. Glucose surges raise insulin and likely are involved in the intense preference for carbohydrate rich foods in ASD patients when plasma glucose is decreasing during times of reduced carbohydrate intake. Interestingly, high-fat diets blunt adrenergic signaling in adipocytes [110]. Older children with ASD show higher levels of cortisol compared to younger children with ASD as well as their typically developing peers during play [111-112].
Allergens and Toxins
It is difficult to interpret abnormal xenobiotic levels in ASD. For instance, a high burden of aluminum, cadmium, lead, mercury, and arsenic was found in a subgroup of a sample of over 500 patients with ASD (113). Other studies have described decreased levels of some of these heavy metals in urine and in hair samples, which may imply that the body is not excreting heavy metals adequately (114). A systematic review of toxicant-related studies in ASD found that pesticides, phthalates, PCBs, solvents, toxic waste sites, air pollutants, and heavy metals were implicated in ASD, with the strongest evidence found for air pollutants and pesticides (115).
Adrenaline infused during a histamine challenge demonstrated higher amounts of histamine required for its known effects on specific airway conductance [118]. This suggests adrenaline to act as protective agent in histamine induced changes. In fact, adrenaline is the therapy of choice in the United States for anaphylactic reactions with an estimated 200,000 people requiring emergency medical care for allergic reactions to food every year [119]. Since plasma allergen exposure in ASD is partly due to dietary factors, genetics and glutathione (GSH) imbalances, it is important to remember the body’s natural response to elevated plasma allergens or anaphylaxis is to release adrenaline.

Conclusion
We propose adrenochrome and adrenolutin to be the critical etiologic basis for the core symptoms observed and described in ASD. The psychomimetic properties of adrenochrome and adrenolutin are well documented in the literature. We propose their involvement with aberrant dopamine signaling in the prefrontal cortex and striatum due to a wide variety of predisposing elements, e.g. genetic polymorphisms as well as dietary and environmental factors. This can have important implications for diagnosis, treatment, and prognosis, as well as for developing fundamental etiological models of these disorders. Further research should focus on the quantification of these metabolites in ASD as well as neurotypical controls.
References
[1] Heacock, R. A. (1959). Chemistry of Adrenochrome And Related Compounds. Chemical Reviews, 59(2), 181–237.
[2] Alberto Bindoli, Maria Pia Rigobello, Lauro Galzigna. Toxicity of aminochromes. Toxicology Letters 1989, 48 (1), 3-20.
[3] Alberto Bindoli, Maria Pia Rigobello, David J. Deeble. Biochemical and toxicological properties of the oxidation products of catecholamines. Free Radical Biology and Medicine 1992, 13 (4), 391-405
[4] R.A. Heacock, W.S. Powell. 6 Adrenochrome and Related Compounds. 1973, 275-340.
[5] Wink DA, Cook JA, Pacelli R, et al. The effect of various nitric oxide-donor agents on hydrogen peroxide-mediated toxicity: a direct correlation between nitric oxide formation and protection. Arch Biochem Biophys. 1996;331(2):241-8.
[6] Daveu, C., Servy, C., Dendane, M., Marin, P., Ducrocq, C., 1997. Oxidation and nitration of catecholamines by nitrogen oxides derived from nitric oxide. Nitric Oxide 1, 234–243.
[7] Yoshie, Y. and Ohshima, H. (1997) Synergistic induction of DNA strand breakage caused by nitric oxide together with catecholamines: implication for neurodegenerative disease. Chem. Res. Toxicol. 10, 1015-1022.
[8] Heacock, R., The Chemistry of Adrenochrome and Related Compounds, 1959
[9] Hoffer, A. (1998). Vitamin B-3 schizophrenia discovery recovery controversy. Kingston, ON: Quarry Books.
[10] Hoffer, A. (1962) – The Effect of Adrenochrome and Adrenolutin On the Behavior of Animals and the Psychology of Man – International Review of Neurobiology Volume 4 International Review of Neurobiology
[11] Schwarz, B.E., Sem-Jacobsen, C.W., Petersen, M.C., 1956. Effects of mescaline, LSD-25, and adrenochrome on depth electrograms in man. AMA Arch. Neurol. Psychiatry 75 (6), 579–587.
[12] Grof, S., Vojtechovsky, M., Vitek, V., Prankova, S., 1963. Clinical and experimental study of central effects of adrenochrome. J. Neuropsychiatr. 4, 33–50
[13] Leach, B.E., and Heath, R.G.: The in vitro oxidation of Epinephrine in Plasma, A.M.A. Arch.Neurol.&Psychiat.76:444-450.1956
[14] Green DE, Richter D. Adrenaline and adrenochrome. Biochem J. 1937;31(4):596-616.
[15] Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247(10):3170-5.
[16] V.M. Costa,R. Silva, L.M Ferriera, P. S. Branco, F. Carvalho, M.L. Bastos,R. A. Carvalho, M.Carvalho, F. Remiao. Oxidation process of adrenaline in freshly isolated rat cardiomyocytes: Formation of adrenochrome, quinoproteins and GSH adduct. Chem Res toxicol. 20 (2007); 1183-1191.
[17] Harley-Mason,J. (1948) Experientia 4,307-308.
[18] Lund,A. (1949)ActaPharmacal.Toxicol.5,75-93.
[19] Fischer,P.andBacq,Z.M. (1949)Cornpt.Rend. Soc. BioI. 143,1159-1161
[20] Bu’Lock,J.D.andHarley-Mason,J. (1951) J. Chern. Soc.2248-2252.1]
[21] Heacock,R.A. and Mattock,G.L.(1963) Can. J. Chern. 41,139-147.12 1
[22] Heacock,RA., Mattock,G.L. and Wilson, D.L. (1963) Can.J.Biochern.Physiol.41,1721-1725.
[23] Krstenansky JL, Xu D, Leitzke R, Saldivar A, Gevorkian R, Shi WX. Adrenochrome and related oxidative metabolites of catecholamines: effects on dopamine neurons and receptor binding profiles. Schizophr Res. 2011;133(1-3):264-5.
[24] Uys MM, Shahid M, Harvey BH. Therapeutic Potential of Selectively Targeting the α-Adrenoceptor in Cognition, Depression, and Schizophrenia-New Developments and Future Perspective. Front Psychiatry. 2017;8:144.
[25] Bücheler MM, Hadamek K, Hein L. Two alpha(2)-adrenergic receptor subtypes, alpha(2A) and alpha(2C), inhibit transmitter release in the brain of gene-targeted mice. Neuroscience. 2002;109(4):819-26.
[26] Fagerholm V, Rokka J, Nyman L, et al. Autographic characterization of a2Cadrenoceptors in the human striatum. Synapse. 2008;62:508–515.
[27] Fremeau, R.T. Jr., Auteliano, D.J., Blum, M., Wilcox, J. and Roberts, J.L., Intervening sequence-specific in situ hybridization: detection of the pro-opiomelanocortin gene primary transcript in individual neurons, Mol. Brain Res., 6 (1989) 197-201.
[28] Palay, S,L. and Chan-Palay, V., Cerebellar Cortex, Cytology and Organization, Springer, Berlin, 1974, 348 pp.
[29] Redmond, D.E. and Huang, Y.tI., New evidence for a locus coeruleus norepinephrine connection with anxiety, Life Sci., 25 (1979) 2149-2162.
[30] Finnema SJ, Hughes ZA, Haaparanta-Solin M, Stepanov V, Nakao R, Varnäs K, et al. Amphetamine decreases α(2C)-adrenoceptor binding of [(11)C]ORM13070: a PET study in the primate brain. Int J Neuropsychopharmacol (2015)
[31] Berridge CW, Spencer RC. Differential cognitive actions of norepinephrine a2 and a1 receptor signaling in the prefrontal cortex. Brain Res. 2016;1641(Pt B):189-96.
[32] Rosin, D.L., Talley, E.M., Lee, A., Stornetta, R.L., Gaylinn, B.D., Guyenet, P.G., Lynch, K.R., 1996. Distribution of alpha 2C-adrenergic receptor-like immunoreactivity in the rat central nervous system. J. Comp. Neurol. 372 (1), 135–165
[33] Inyushin MU, Arencibia-albite F, Vázquez-torres R, Vélez-hernández ME, Jiménez-rivera CA. Alpha-2 noradrenergic receptor activation inhibits the hyperpolarization-activated cation current (Ih) in neurons of the ventral tegmental area. Neuroscience. 2010;167(2):287-97.
[34] Yamamoto K, Hornykiewicz O. Proposal for a noradrenaline hypothesis of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2004) 28:913–22.doi:10.1016/j.pnpbp.2004.05.033
[35] Sallinen J, Höglund I, Engström M, Lehtimäki J, Virtanen R, Sirviö J, et al. Pharmacological characterization and CNS effects of a novel highly selective α2C-adrenoceptor antagonist JP-1302. Br J Pharmacol (2007) 150:391–402.
[36] Sallinen J, Holappa J, Koivisto A, Kuokkanen K, Chapman H, Lehtimäki J, et al. Pharmacological characterisation of a structurally novel α2Cadrenoceptor antagonist ORM-10921 and its effects in neuropsychiatric models. Basic Clin Pharmacol Toxicol (2013) 113:239–49.
[37] Sallinen J, Rouru J, Lehtimäki J, Marjamaeki P, Haaparanta-Solin M, Arponen E, et al. ORM-12741: receptor pharmacology of a novel alpha2c- adrenergic receptor subtype selective antagonist with multi-therapeutic potential. Neuropsychopharmacology (2013)
[38] Simpson EH, Kellendonk C, Kandel E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron. 2010;65(5):585-96. 1
[39] Lipton DM, Gonzales BJ, Citri A. Dorsal Striatal Circuits for Habits, Compulsions and Addictions. Front Syst Neurosci. 2019;13:28.
[40] Starke K. (2001) Presynaptic autoreceptors in the third decade: focus on α2‐adrenoceptors. J. Neurochem. 78, 685–693.
[41] Lähdesmäki J., Sallinen J., MacDonald E., Sirviö J. and Scheinin M. (2003) α2‐Adrenergic drug effects on brain monoamines, locomotion, and body temperature are largely abolished in mice lacking the α2A‐adrenoceptor subtype. Neuropharmacology 44, 882–892.
[42] Inyushin MU, Arencibia-albite F, Vázquez-torres R, Vélez-hernández ME, Jiménez-rivera CA. Alpha-2 noradrenergic receptor activation inhibits the hyperpolarization-activated cation current (Ih) in neurons of the ventral tegmental area. Neuroscience. 2010;167(2):287-97.
[43] Bleuler E. Dementia Precox oder Gruppe der Schizophrenien. In: Handbuch
der Psychiatrie. Leipzig, Germany: Deuticke; 1911.
[44] 7. Crespi BJ. Revisiting Bleuler: relationship between autism and schizophrenia.
Br J Psychiatry. 2010;196(6):495. author reply 495-496.
[45] Kanner L. Autistic disturbances of affective contact. Acta Paedopsychiatr.
1968;35(4):100–36.
[46] Parnas J, Bovet P. Autism in schizophrenia revisited. Compr Psychiatry.
1991;32(1):7–21.
[47] Kästner A, Begemann M, Michel TM, et al. Autism beyond diagnostic categories: characterization of autistic phenotypes in schizophrenia. BMC Psychiatry. 2015;15:115.
[48] Lake CR, Ziegler MG, Murphy DL. Increased norepinephrine levels and decreased dopamine-beta-hydroxylase activity in primary autism. Arch Gen Psychiatry. 1977;34(5):553-6.
[49] Israngkun PP, Newman HA, Patel ST, Duruibe VA, Abou-issa H. Potential biochemical markers for infantile autism. Neurochem Pathol. 1986;5(1):51-70.
[50] Launay JM, Bursztejn C, Ferrari P, et al. Catecholamines metabolism in infantile autism: a controlled study of 22 autistic children. J Autism Dev Disord. 1987;17(3):333-47.
[51] Barthelemy C, Bruneau N, Cottet-eymard JM, et al. Urinary free and conjugated catecholamines and metabolites in autistic children. J Autism Dev Disord. 1988;18(4):583-91.
[52] Hérault J, Martineau J, Perrot-beaugerie A, et al. Investigation of whole blood and urine monoamines in autism. Eur Child Adolesc Psychiatry. 1993;2(4):211-220.
[53] Breier A, Wolkowitz OM, Roy A, Potter WZ, Pickar D. Plasma norepinephrine in chronic schizophrenia. Am J Psychiatry. 1990;147(11):1467-70.
[54] Lake CR, Sternberg DE, Van kammen DP, et al. Schizophrenia: elevated cerebrospinal fluid norepinephrine. Science. 1980;207(4428):331-3.
[55] Farley IJ, Price KS, Mccullough E, Deck JH, Hordynski W, Hornykiewicz O. Norepinephrine in chronic paranoid schizophrenia: above-normal levels in limbic forebrain. Science. 1978;200(4340):456-8. 1
[56] Chisholm K, Lin A, Abu-akel A, Wood SJ. The association between autism and schizophrenia spectrum disorders: A review of eight alternate models of co-occurrence. Neurosci Biobehav Rev. 2015;55:173-83.
[57] Kästner A, Begemann M, Michel TM, et al. Autism beyond diagnostic categories: characterization of autistic phenotypes in schizophrenia. BMC Psychiatry. 2015;15:115.
[58] Cirillo MA, Seidman LJ. Verbal declarative memory dysfunction in schizophrenia: from clinical assessment to genetics and brain mechanisms. Neuropsychology Review. 2003;13:43–77.
[59] Goldman-rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6(4):348-57.
[60] Southwick JS, Bigler ED, Froehlich A, et al. Memory functioning in children and adolescents with autism. Neuropsychology. 2011;25(6):702-710.
[61] Berenguer C, Roselló B, Colomer C, Baixauli I, Miranda A. Children with autism and attention deficit hyperactivity disorder. Relationships between symptoms and executive function, theory of mind, and behavioral problems. Res Dev Disabil. 2018;83:260-269.
[62] Allen G, Courchesne E. Attention function and dysfunction in autism. Front Biosci. 2001;6:D105-19.
[63] Wang Y, Zhang YB, Liu LL, et al. A Meta-Analysis of Working Memory Impairments in Autism Spectrum Disorders. Neuropsychol Rev. 2017;27(1):46-61.
[64] Corbett BA, Mendoza S, Abdullah M, Wegelin JA, Levine S. Cortisol circadian rhythms and
response to stress in children with autism. Psychoneuroendocrinology. 2006 Jan; 31(1):59–68.
[65] Tordjman S, Anderson GM, McBride PA, Hertzig ME, Snow ME, Hall LM, et al. Plasma beta endorphin, adrenocorticotropin hormone, and cortisol in autism. J Child Psychol Psychiatry. 1997
Sep; 38(6):705–15.
[66] Mishra, H. P., and Fridovich, I. (1972) The role of superoxide anion in the autoxidation of epinephrine and a single assay for superoxide dismutase. J. Biol. Chem. 247, 3170–3175.
[67] Alhasan R, Njus D. The epinephrine assay for superoxide: why dopamine does not work. Anal Biochem. 2008;381(1):142-7.
[68] Cekici H, Sanlier N. Current nutritional approaches in managing autism spectrum disorder: A review. Nutr Neurosci. 2019;22(3):145-155.
[69] Shaw W, Kassen E, Chaves E. Increased urinary excretion of analogs of Krebs cycle metabolites and arabinose in two brothers with autistic features. Clin Chem 1995;41:1094–104.
[70] Shaw W, Kassen E, Chaves E. Assessment of antifungal drug therapy in autism by measurement of suspected microbial metabolites in urine with gas chromatography-mass spectrometry. Clin Pract Alter Med 2000;1:15–26.
[71] Kaluzna-Czaplinska J, Błaszczyk S. The level of arabinitol in autistic children after probiotic therapy. Nutrition 2012;28(2): 124–6 1
[72] Reichelt KL, Knivsberg AM. The possibility and probability of a gut-to-brain connection in autism. Ann Clin Psychiatry 2009; 21:205–11
[73] Shaw W. Increased urinary excretion of a 3-(3-hydroxyphenyl)- 3-hydroxypropionic acid (HPHPA), an abnormal phenylalanine metabolite of Clostridia spp. in the gastrointestinal tract, in urine samples from patients with autism and schizophrenia. Nutr Neurosci 2010;13:135–43.
[74] Reichelt KL, Knivsberg AM. Can the pathophysiology of autism be explained by the nature of the discovered urine peptides? Nutr Neurosci 2003;6:19–28.
[75] Hopper DG, Bolton VE, Guilford FT, Straus DC. Mycotoxin detection in human samples from patients exposed to environmental molds. Int J Sci 2009;10:1465–75
[76] Yap IK, Angley M, Veselkov KA, Holmes E, Lindon JC, Nicholson JK. Urinary metabolic phenotyping differentiates children with autism from their unaffected siblings and agematched controls. J Proteome Res 2010;9:2996–3004.
[77] Clayton TA. Metabolic differences underlying two distinct rat urinary phenotypes, a suggested role for gut microbial metabolism of phenylalanine and a possible connection to autism. FEBS Lett 2012;586:956–61.
[78] Emanuele E, Orsi P, Boso M, et al. Low-grade endotoxemia in patients with severe autism. Neurosci Lett. 2010;471(3):162-5.
[79] S.J. James, S. Melnyk, S. Jernigan, M.A. Cleves, C.H. Halsted, D.H. Wong, P. Cutler, K.
Bock, M. Boris, J.J. Bradstreet, Metabolic endophenotype and related genotypes are associated with
oxidative stress in children with autism, Am. J. Med. Genet. 141(8) (2006) 947-956.
[80] D.A. Rossignol, R.E. Frye, Evidence linking oxidative stress, mitochondrial dysfunction, and
inflammation in the brain of individuals with autism, Front. Physiol. 5 (2014) 150.
[81] A. Ghezzo, P. Visconti, P.M. Abruzzo, A. Bolotta, C. Ferreri, G. Gobbi, G. Malisardi, S.
Manfredini, M. Marini, L. Nanetti, Oxidative stress and erythrocyte membrane alterations in
children with autism: correlation with clinical features, PLoS One 8(6) (2013) e66418.
[82] K. Yui, N. Tanuma, H. Yamada, Y. Kawasaki, Reduced endogenous urinary total antioxidant
power and its relation of plasma antioxidant activity of superoxide dismutase in individuals with
autism spectrum disorder, Int. J. Dev. Neurosci. 60 (2017) 70-77.
[83] E.M. Sajdel-Sulkowska, M. Xu, N. Koibuchi, Increase in cerebellar neurotrophin-3 and
oxidative stress markers in autism, Cerebellum 8(3) (2009) 366-372.
[84] T. Evans, S. Siedlak, L. Lu, X. Fu, Z. Wang, W. McGinnis, E. Fakhoury, R. Castellani, S.
Hazen, W. Walsh, The autistic phenotype exhibits a remarkably localized modification of brain
protein by products of free radical-induced lipid oxidation, Am. J. Biochem. Biotechnol. 4(2)
(2008) 61-72.
[85] E. Sajdel-Sulkowska, B. Lipinski, H. Windom, T. Audhya, W. McGinnis, Oxidative stress in
autism: elevated cerebellar 3-nitrotyrosine levels, Am. J. Biochem. Biotechnol. 4(2) (2008) 73-84.
[86] G. Bjørklund, N.A. Meguid, M. A el-bana, A.A. Tinkov, K. Saad, M. Dadar, M. Hemimi, A. 1
Skalny, B. Hosnedlová, R. Kizek, J. Osredkar, M.A. Urbina, T. Fabjan, A. El-Houfey, J. Kałużna-
Czaplińska, P. Gątarek, S. Chirumbolo, Oxidative stress in autism spectrum disorder, Mol.
Neurobiol. 57 (2020) 2314–2332.
[87] G. Tang, P.G. Rios, S.-H. Kuo, H.O. Akman, G. Rosoklija, K. Tanji, A. Dwork, E.A. Schon, S.
DiMauro, J. Goldman, Mitochondrial abnormalities in temporal lobe of autistic brain, Neurobiol.
Dis. 54 (2013) 349-361.
[88] F. Hollis, A.K. Kanellopoulos, C. Bagni, Mitochondrial dysfunction in Autism Spectrum
Disorder: clinical features and perspectives, Curr. Opin. Neurobiol. 45 (2017) 178-187.
[89] L.J. Raymond, R.C. Deth, N.V. Ralston, Potential role of selenoenzymes and antioxidant
metabolism in relation to autism etiology and pathology, Autism Res. Treat. 2014 (2014).
[90] A. Ranjbar, V. Rashedi, M. Rezaei, Comparison of urinary oxidative biomarkers in Iranian
children with autism, Res. Dev. Disabil. 35(11) (2014) 2751-2755.
[91] A. Strunecka, O. Strunecky, Chronic Fluoride Exposure and the Risk of Autism Spectrum
Disorder, Int. J. Environ. Res. Public Health 16(18) (2019) 3431.
[92] E. Mousavinejad, M.A. Ghaffari, F. Riahi, M. Hajmohammadi, Z. Tiznobeyk, M.
Mousavinejad, Coenzyme Q10 supplementation reduces oxidative stress and decreases antioxidant
enzyme activity in children with autism spectrum disorders, Psychiatry Res, 2018, pp. 62-69.
[93] Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012;17(4):389-401.
[94] G. Morris, G. Anderson, O. Dean, M. Berk, P. Galecki, M. Martin-Subero, M. Maes, The
glutathione system: a new drug target in neuroimmune disorders, Mol. Neurobiol. 50(3) (2014)
1059-1084.
[95] M. Mari, A. Morales, A. Colell, C. García-Ruiz, J.C. Fernández-Checa, Mitochondrial
glutathione, a key survival antioxidant, Antioxid. Redox Signal. 11(11) (2009) 2685-2700.
[96] C.B. Pocernich, D.A. Butterfield, Elevation of glutathione as a therapeutic strategy in
Alzheimer disease, Biochim. Biophys. Acta Mol. Basis Dis. 1822(5) (2012) 625-630.
[97] M. Mari, A. Morales, A. Colell, C. García-Ruiz, J.C. Fernández-Checa, Mitochondrial
glutathione, a key survival antioxidant, Antioxid. Redox Signal. 11(11) (2009) 2685-2700.
[98] J.B. Adams, T. Audhya, S. McDonough-Means, R.A. Rubin, D. Quig, E. Geis, E. Gehn, M.
Loresto, J. Mitchell, S. Atwood, Nutritional and metabolic status of children with autism vs.
neurotypical children, and the association with autism severity, Nutr. Metab. 8(1) (2011) 34.
[99] Y.A. Al-Yafee, L.Y. Al-Ayadhi, S.H. Haq, A.K. El-Ansary, Novel metabolic biomarkers
related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia, BMC
Neurol. 11(1) (2011) 139. 1
[100] S.J. James, S. Rose, S. Melnyk, S. Jernigan, S. Blossom, O. Pavliv, D.W. Gaylor, Cellular and
mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with
autism, FASEB J. 23(8) (2009) 2374-2383
[101] Afrazeh M, Saedisar S, Khakzad MR, Hojati M. Measurement of Serum Superoxide Dismutase and Its Relevance to Disease Intensity Autistic Children. Maedica (Buchar). 2015;10(4):315-318.
[102] Buie T, Campbell DB, Fuchs GJ 3rd, et al. Evaluation, diagnosis, and treatment of
gastrointestinal disorders in individuals with ASDs: a consensus report. Pediatrics
2010;125(Suppl 1):S1–18.
[103] McElhanon BO, McCracken C, Karpen S, et al. Gastrointestinal symptoms in autism spectrum disorder: a meta-analysis. Pediatrics 2014;133(5):872–83.
[104] Marí-Bauset S, Zazpe I, Mari-Sanchis A, Llopis-González A, Morales-Suárez-Varela M. Food selectivity in autism spectrum disorders: a systematic review. J Child Neurol 2013;29:1554–61. 23.
[105] Ranjan S, Nasser J a. Nutritional status of individuals with autism spectrum disorders: do we know enough? Adv Nutr 2015;15;6(4):397–407
[106] Esteban-Figueroa P, Canals J, Fernández JC, Arija val V. Differences in food consumption and nutritional intake between children with autism spectrum disorders and typically developing children: A meta-analysis. Autism. 2019;23(5):1079-1095.
[107] Hamburg S, Hendler R, Sherwin RS. Influence of small increments of epinephrine on glucose tolerance in normal humans. Ann Intern Med. 1980;93(4):566-8.
[108] Stern M. Insulin signaling and autism. Front Endocrinol (Lausanne). 2011;2:54.
[109] Ross AP, Darling JN, Parent MB. Excess intake of fat and sugar potentiates epinephrine-induced hyperglycemia in male rats. J Diabetes Complicat. 2015;29(3):329-37.
[110] Gaidhu, M. P., Anthony, N. M., Patel, P., Hawke, T. J., & Ceddia, R. B. (2010). Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by highfat diet: role of ATGL, HSL, and AMPK. American Journal of Physiology. Cell Physiology, 298, C961–C971
[111] Corbett BA, Schupp CW, Lanni KE. Comparing biobehavioral profiles across two social stress paradigms in children with and without autism spectrum disorders. Mol Autism. 2012 Nov 17.3(1):13.
[112] Schupp CW, Simon DS, Corbett BA. Cortisol responsivity differences in children with autism
spectrum disorders during free and cooperative play. Journal of Autism and Developmental
Disorders. in press.
[113] Yasuda H, Yasuda Y, Tsutsui T. Estimation of autistic children by metallomics analysis. Sci Rep (2013) 3:1199.10.1038/srep01199
[114] Wang L, Angley MT, Gerber JP, Sorich MJ. A review of candidate urinary biomarkers for autism spectrum disorder. Biomarkers (2011) 16:537–5210.3109/1354750X.2011.598564 1
[115] Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Transl Psychiatry. 2014;4:e360.
[116] Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatr. 2004;113:e472–e486.
[117] Keller F, Persico AM. The neurobiological context of autism. Mol Neurobiol. 2003;28:1–22.
[118] Warren JB, Dalton N, Turner C, Clark TJ. Protective effect of circulating epinephrine within the physiologic range on the airway response to inhaled histamine in nonasthmatic subjects. J Allergy Clin Immunol. 1984;74(5):683-6.
[119] 6 Clark S, Espinola J, Rudders SA, Banerji, A, Camargo CA. Frequency of US emergency department visits for food-related acute allergic reactions. J Allergy Clin Immunol. 2011; 127(3):682-683
[120] Sasaki M, Nakagawa E, Sugai K, Shimizu Y, Hattori A, Nonoda Y, et al. Brain perfusion SPECT and EEG findings in children with autism spectrum disorders and medically intractable epilepsy. Brain Dev. 2010;32:776–82.
[121] Ohnishi T, Matsuda H, Hashimoto T, et al. Abnormal regional cerebral blood flow in childhood autism. Brain. 2000;123 ( Pt 9):1838-44.
[122] Wilcox J, Tsuang MT, Ledger E, Algeo J, Schnurr T. Brain perfusion in autism varies with age. Neuropsychobiology. 2002;46(1):13-6.
[123] Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 6: Widely Projecting Systems: Monoamines, Acetylcholine, and Orexin”. In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. pp. 147–148, 154–157. ISBN 9780071481274.
[124] Felger JC, Li Z, Haroon E, et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. 2016;21(10):1358-65.
[125] Brown AS, Sourander A, Hinkka-yli-salomäki S, Mckeague IW, Sundvall J, Surcel HM. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry. 2014;19(2):259-64.
[126] Aronson JK. “Where name and image meet”–the argument for “adrenaline”. BMJ. 2000;320(7233):506-9.
[127] Barnett JH, Jones PB, Robbins TW, et al. Effects of the catechol-O-methyltransferase Val158Met polymorphism on executive function: a meta-analysis of the Wisconsin Card Sort Test in schizophrenia and healthy controls. Mol Psychiatry 2007; 12: 502–509.
[128] Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nature Reviews Neuroscience. 2006;7:818–827.
[129] James SJ, Melnyk S, Jernigan S, et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(8):947-56.
[130] Sadeghiyeh T, Dastgheib SA, Mirzaee-khoramabadi K, et al. Association of MTHFR 677C>T and 1298A>C polymorphisms with susceptibility to autism: A systematic review and meta-analysis. Asian J Psychiatr. 2019;46:54-61.
[131] Supekar K, Kochalka J, Schaer M, et al. Deficits in mesolimbic reward pathway underlie social interaction impairments in children with autism. Brain. 2018;141(9):2795-2805. 1
[132] Oliver G, Schafer EA. The physiological effects of extracts of the suprarenal capsules. J Physiol 1895; 18:230-76.
[133] Cengiz M, Cezayirli E, Bayoglu B, Asliyuksek H, Kocabasoglu N. Catechol-O-Methyltransferase Val158Met and brain-derived neurotrophic factor Val66Met gene polymorphisms in paraphilic sexual offenders. Indian J Psychiatry. 2019;61(3):253-257.
[134] Kabukcu Basay B, Buber A, Basay O, Alacam H, Ozturk O, Suren S, et al. White matter alterations related to attention-deficit hyperactivity disorder and COMT val (158) met polymorphism: Children with valine homozygote attention-deficit hyperactivity disorder have altered white matter connectivity in the right cingulum (cingulate gyrus) Neuropsychiatr Dis Treat. 2016;12:969–81. [PMC free article] [PubMed] [Google Scholar]
[135] Pełka-Wysiecka J, Wroński M, Jasiewicz A, Grzywacz A, Tybura P, Kucharska-Mazur J, et al. BDNF rs 6265 polymorphism and COMT rs 4680 polymorphism in deficit schizophrenia in polish sample. Pharmacol Rep. 2013;65:1185–93. [PubMed] [Google Scholar]
[136] Cengiz M, Okutan SN, Bayoglu B, Sakalli Kani A, Bayar R, Kocabasoglu N, et al. Genetic polymorphism of the serotonin transporter gene, SLC6A4 rs16965628, is associated with obsessive compulsive disorder. Genet Test Mol Biomarkers. 2015;19:228–34. [PubMed] [Google Scholar]
[137] Muellner J, Gharrad I, Habert MO, Kas A, Martini JB, Cormier-Dequaire F, et al. Dopaminergic denervation severity depends on COMT val158Met polymorphism in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21:471–6. [PubMed] [Google Scholar]
[138] Sawa A, Snyder SH. Schizophrenia: diverse approaches to a complex disease. Science. 2002;296(5568):692-5.
[139] Gogos, J.A., Morgan, M., Luine, V., Santha, M., Ogawa, S., Pfaff, D., and Karayiorgou, M. (1998). Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proceedings of the National Academy of Sciences, 95(17), 9991-96.
[140] https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=201.323
[141] McFarland G, La Joie E, Thomas P, Lyons-Weiler J. Acute exposure and chronic retention of aluminum in three vaccine schedules and effects of genetic and environmental variation. Journal of Trace Elements in Medicine and Biology. 2020;58:126444.
[142] Mold M, Umar D, King A, Exley C. Aluminium in brain tissue in autism. Journal of Trace Elements in Medicine and Biology. 2018;46:76-82